Weighted samples: the Samples, MCMCSamples, and NestedSamples data frames
The anesthetic.samples.Samples data frame at its core is a
pandas.DataFrame, and as such comes with all the pandas functionality.
The anesthetic.samples.MCMCSamples and
anesthetic.samples.NestedSamples data frames share all the
functionality of the more general Samples class, but come with some
additional MCMC or nested sampling specific functionality.
from anesthetic import read_chains, make_2d_axes
samples = read_chains("../../tests/example_data/pc_250")
General weighted sample functionality
The important extension to the pandas.Series and
pandas.DataFrame classes, is that in anesthetic the data frames are
weighted (see anesthetic.weighted_pandas.WeightedSeries and
anesthetic.weighted_pandas.WeightedDataFrame), where the weights form
part of a pandas.MultiIndex and are therefore retained when slicing.
Mean, standard deviation, median, quantiles, etc.
The weights are automatically taken into account in summary statistics such as mean, variance, covariance, skewness, kurtosis, standard deviation, median, quantiles, etc.
x0_median = samples.x0.median()
x1_mean = samples.x1.mean()
x1_std = samples.x1.std()
x2_min = samples.x2.min()
x2_95percentile = samples.x2.quantile(q=0.95)
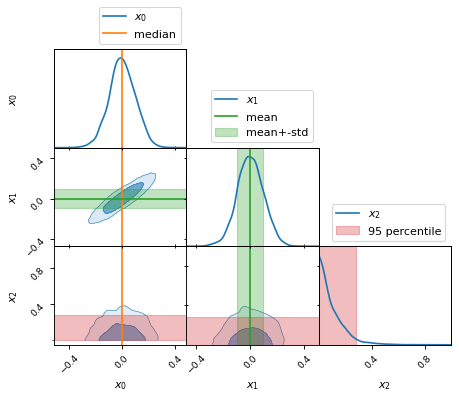
fig, axes = make_2d_axes(['x0', 'x1', 'x2'], upper=False)
samples.plot_2d(axes, label=None)
axes.axlines({'x0': x0_median}, c='C1', label="median")
axes.axlines({'x1': x1_mean}, c='C2', label="mean")
axes.axspans({'x1': (x1_mean-x1_std, x1_mean+x1_std)}, c='C2', alpha=0.3, label="mean+-std")
axes.axspans({'x2': (x2_min, x2_95percentile)}, c='C3', alpha=0.3, label="95 percentile")
axes.iloc[0, 0].legend(bbox_to_anchor=(1, 1), loc='lower right')
axes.iloc[1, 1].legend(bbox_to_anchor=(1, 1), loc='lower right')
axes.iloc[2, 2].legend(bbox_to_anchor=(1, 1), loc='lower right')
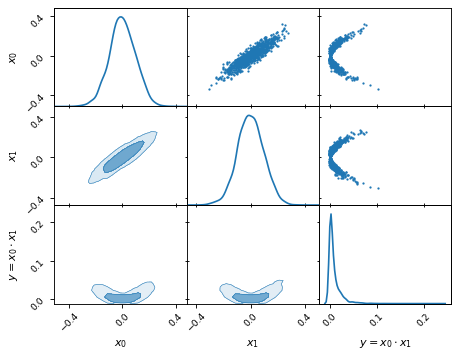
Creating new from existing columns
We can define new parameters with relative ease. For example, given two
parameters x0 and x1, we can compute the derived parameter
y = x0 * x1:
samples['y'] = samples['x1'] * samples['x0']
samples.set_label('y', '$y=x_0 \\cdot x_1$')
samples.plot_2d(['x0', 'x1', 'y'])
MCMC statistics
Markov Chain Monte Carlo (short MCMC) samples as the name states come from Markov chains, and as such come with some MCMC specific properties and potential issues, e.g. correlation of successive steps, a burn-in phase, or questions of convergence.
We have an example data set (at the relative path
anesthetic/tests/example_data/ with the file root cb) that emphasizes
potential MCMC issues. Note, while this was run with Cobaya, we had to actually put in some
effort to make Cobaya produce such a bad burn-in stage. With its usual
optimisation settings it normally produces much better results.
Chains
When MCMC data is read in, anesthetic automatically keeps track of multiple
chains that were run in parallel via the 'chain' parameter. You can split
the chains into separate samples via the pandas.DataFrame.groupby()
method:
from anesthetic import read_chains, make_2d_axes
mcmc_samples = read_chains("../../tests/example_data/cb")
chains = mcmc_samples.groupby(('chain', '$n_\\mathrm{chain}$'), group_keys=False)
chain1 = chains.get_group(1)
chain2 = chains.get_group(2).reset_index(drop=True)
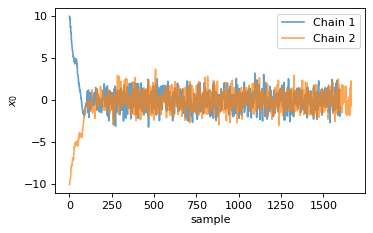
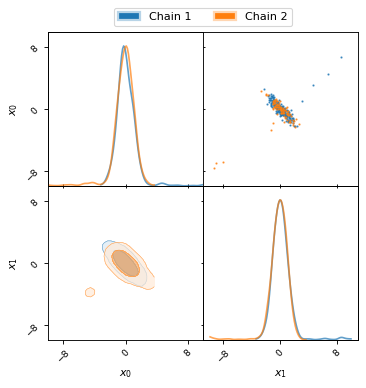
For this example MCMC run the initial burn-in phase is very apparent, as can be seen in the following two plots.
fig, ax = plt.subplots(figsize=(5, 3))
ax = chain1.x0.plot.line(alpha=0.7, label="Chain 1")
ax = chain2.x0.plot.line(alpha=0.7, label="Chain 2")
ax.set_ylabel(chain1.get_label('x0'))
ax.set_xlabel("sample")
ax.legend()
fig, axes = make_2d_axes(['x0', 'x1'], figsize=(5, 5))
chain1.plot_2d(axes, alpha=0.7, label="Chain 1")
chain2.plot_2d(axes, alpha=0.7, label="Chain 2")
axes.iloc[-1, 0].legend(bbox_to_anchor=(len(axes)/2, len(axes)), loc='lower center', ncol=2)
Remove burn-in
To get rid of the initial burn-in phase, you can use the
anesthetic.samples.MCMCSamples.remove_burn_in() method:
mcmc_burnout = mcmc_samples.remove_burn_in(burn_in=0.1)
Positive burn_in values are interpreted as the first samples to
remove, whereas negative burn_in values are interpreted as the last
samples to keep. You can think of it in the usual python slicing mentality:
samples[burn_in:].
If 0 < abs(burn_in) < 1 then it is interpreted as a fraction of the total
number of samples in the respective chain.
To see how remove_burn_in has removed the burn-in samples in both
chains, see the plot in the following section, alongside an assessment of
convergence.
Gelman–Rubin statistic
Another important issue when it comes to MCMC samples is assessing convergence. In anesthetic we have implemented the modified Gelman–Rubin statistic as described in Antony Lewis (2013). For the underlying (more theoretical) accounts of this statistic, see e.g. Gelman and Rubin (1992) and Brooks and Gelman (1998).
Provided you have an MCMC run containing multiple chains, you can compute the
Gelman–Rubin R-1 statistic using the
anesthetic.samples.MCMCSamples.Gelman_Rubin() method:
Rminus1_old = mcmc_samples.Gelman_Rubin()
Rminus1_new = mcmc_burnout.Gelman_Rubin()
Rminus1_par = mcmc_burnout.Gelman_Rubin(per_param='par')
You can get the convergence per parameter by passing the keyword
per_param='par'. By passing per_param='cov' you will even get the
covariant part of the convergence of pairs of parameters.
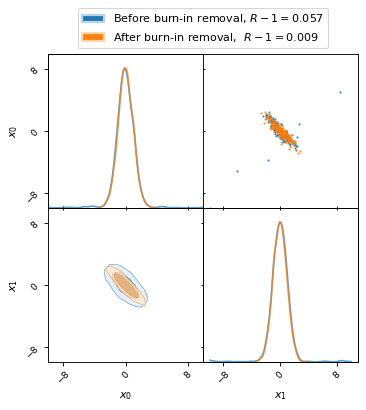
The following plot shows how remove_burn_in gets rid of burn-in samples.
Note the stark difference in the Gelman–Rubin statistic, as listed in the
legend, depending on whether burn-in samples were removed or not.
fig, axes = make_2d_axes(['x0', 'x1'], figsize=(5, 5))
mcmc_samples.plot_2d(axes, alpha=0.7, label="Before burn-in removal, $R-1=%.3f$" % Rminus1_old)
mcmc_burnout.plot_2d(axes, alpha=0.7, label="After burn-in removal, $R-1=%.3f$" % Rminus1_new)
axes.iloc[-1, 0].legend(bbox_to_anchor=(len(axes)/2, len(axes)), loc='lower center')
Note
Unless you specify which parameters to compute the Gelman–Rubin statistic
for (by passing the keyword params), anesthetic will use _all_
parameters in the data frame except those containing ‘prior’, ‘chi2’, or
‘logL’ in their name. So if you for example want to exclude derived
parameters, you should pass params directly.
Nested sampling statistics
Anesthetic really comes to the fore for nested sampling (for details on nested sampling we recommend John Skilling, 2006). We can do all of the above and more with the power that nested sampling chains provide.
from anesthetic import read_chains, make_2d_axes
nested_samples = read_chains("../../tests/example_data/pc")
nested_samples['y'] = nested_samples['x1'] * nested_samples['x0']
nested_samples.set_label('y', '$y=x_0 \\cdot x_1$')
Prior distribution
While MCMC explores effectively only the posterior bulk, nested sampling
explores the full parameter space, allowing us to calculate and plot not only
the posterior distribution, but also the prior distribution, which you can get with the anesthetic.samples.NestedSamples.prior() method:
prior_samples = nested_samples.prior()
Note
Note that the .prior() method is really just a shorthand for
.set_beta(beta=0), i.e. for setting the inverse temperature parameter
beta=0 (where 1/beta=kT) in the
anesthetic.samples.NestedSamples.set_beta() method, which allows you to
get the distribution at any temperature.
This allows us to plot both prior and posterior distributions together. Note,
how the prior is also computed for the derived parameter y:
fig, axes = make_2d_axes(['x0', 'x1', 'y'])
prior_samples.plot_2d(axes, label="prior")
nested_samples.plot_2d(axes, label="posterior")
axes.iloc[-1, 0].legend(bbox_to_anchor=(len(axes)/2, len(axes)), loc='lower center', ncol=2)
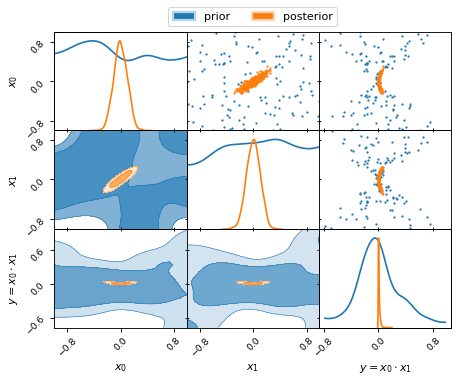
Note, how the uniform priors on the parameters x0 and x1 lead to a
non-uniform prior on the derived parameter y.
Note further the different colour gradient in the posterior contours and the
prior contours. While the iso-probability contour levels are defined by the
amount of probability mass they contain, the colours are assigned according to
the probability density in the contour. As such, the lower probability density
in the posterior tails is reflected in the lighter colour shading of the second
compared to the first contour level. In contrast, the uniform probability
density of the prior distributions of x0 and x1 is reflected in the
similar colour shading of both contour levels.
Bayesian statistics
Thanks to the power of nested sampling, we can compute Bayesian statistics from the nested samples, such as the following:
Bayesian (log-)evidence
anesthetic.samples.NestedSamples.logZ()Kullback–Leibler (KL) divergence
anesthetic.samples.NestedSamples.D_KL()Posterior average of the log-likelihood
anesthetic.samples.NestedSamples.logL_P()
(this connects Bayesian evidence with KL-divergence aslogZ = logL_P - D_KL, allowing the interpretation of the Bayesian evidence as a trade-off between model fitlogL_Pand Occam penaltyD_KL, see also our paper Hergt, Handley, Hobson, and Lasenby (2021))Gaussian model dimensionality
anesthetic.samples.NestedSamples.d_G()
(for more, see our paper Handley and Lemos (2019))All of the above in one go, using
anesthetic.samples.NestedSamples.stats()
By default (i.e. without passing any additional keywords) the mean values for these quantities are computed:
bayesian_means = nested_samples.stats()
Passing an integer number nsamples will create a data frame of samples
reflecting the underlying distributions of the Bayesian statistics:
nsamples = 2000
bayesian_stats = nested_samples.stats(nsamples)
Since bayesian_stats is an instance of anesthetic.samples.Samples,
the same plotting functions can be used as for the posterior plots above.
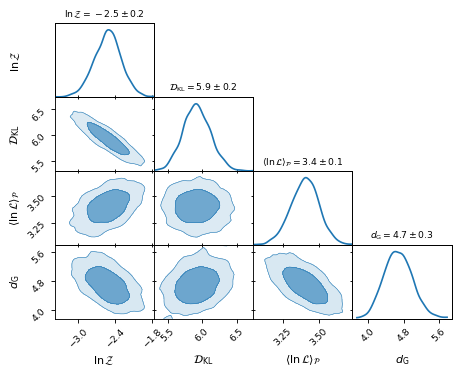
Plotting the 2D distributions allows us to inspect the correlation between the
inferences:
fig, axes = make_2d_axes(['logZ', 'D_KL', 'logL_P', 'd_G'], upper=False)
bayesian_stats.plot_2d(axes);
for y, row in axes.iterrows():
for x, ax in row.items():
if x == y:
ax.set_title("%s$ = %.2g \\pm %.1g$"
% (bayesian_stats.get_label(x),
bayesian_stats[x].mean(),
bayesian_stats[x].std()),
fontsize='small')
Nested Sampling GUI
We can also set up an interactive plot, which allows us to replay a nested sampling run after the fact.
nested_samples.gui()